object(WP_Post)#1486 (24) {
["ID"]=>
int(601)
["post_author"]=>
string(1) "1"
["post_date"]=>
string(19) "2021-12-08 08:02:11"
["post_date_gmt"]=>
string(19) "2021-12-08 08:02:11"
["post_content"]=>
string(575) "Klinikinių tyrimų įmonė UAB „BIO1“ ieško sveikų savanorių, norinčių dalyvauti vaistinių preparatų klinikiniuose tyrimuose. Norėdami gauti informaciją apie planuojamus klinikinius tyrimus – kviečiame registruotis užpildydant žemiau pateiktą anketą. Kreipdamiesi į mus sutinkate gauti informaciją, bet neįsipareigojate dalyvauti klinikiniame tyrime.
[button title="Užpildyti anketą" link="https://forms.office.com/Pages/ResponsePage.aspx?id=t_E9jUInpESiRhrwsmKd6_zq6h21NYpNjkyig6a5AadUQTVFSElCREpQTVNLT1hBMzdUNjRGTlZHVS4u" color="theme" large="1"]"
["post_title"]=>
string(49) "
Kvietimas dalyvauti klinikiniame tyrime "
["post_excerpt"]=>
string(0) ""
["post_status"]=>
string(7) "publish"
["comment_status"]=>
string(4) "open"
["ping_status"]=>
string(4) "open"
["post_password"]=>
string(0) ""
["post_name"]=>
string(39) "kvietimas-dalyvauti-klinikiniame-tyrime"
["to_ping"]=>
string(0) ""
["pinged"]=>
string(0) ""
["post_modified"]=>
string(19) "2022-09-28 12:17:27"
["post_modified_gmt"]=>
string(19) "2022-09-28 12:17:27"
["post_content_filtered"]=>
string(0) ""
["post_parent"]=>
int(0)
["guid"]=>
string(29) "https://bio1trials.com/?p=601"
["menu_order"]=>
int(0)
["post_type"]=>
string(4) "post"
["post_mime_type"]=>
string(0) ""
["comment_count"]=>
string(1) "0"
["filter"]=>
string(3) "raw"
}
Kvietimas dalyvauti klinikiniame tyrime
Klinikinių tyrimų įmonė UAB „BIO1“ ieško sveikų savanorių, norinčių dalyvauti vaistinių preparatų klinikiniuose tyrimuose. Norėdami gauti informaciją apie planuojamus klinikinius tyrimus – kviečiame registruotis užpildydant žemiau […]
Read more >
object(WP_Post)#1485 (24) {
["ID"]=>
int(549)
["post_author"]=>
string(1) "1"
["post_date"]=>
string(19) "2020-11-04 09:53:20"
["post_date_gmt"]=>
string(19) "2020-11-04 09:53:20"
["post_content"]=>
string(5564) "
Quality assurance in early phase clinical trials plays an important role in minimizing potential risk to volunteers or patients who take part in these types of studies and may help to eliminate potential problems before they occur.
The ICH-GCP principles for clinical trials are applied for early phase clinical trials the same, but some specific points that should be considered while auditing or managing early phase units. The three key factors for the successful operations in the early phase units combine professional staff, proper facilities and equipment, and the administration of healthy volunteers.
Professional staff
The first factor leading to success in early phase clinical trials is experienced, trained, and dedicated staff. Indeed, they are key to the safety of volunteers or patients in early phase trials. Proper resourcing and allocation of staff to the trials is an important aspect of the unit’s day to day function.
Enough staff should be available during dosing days for multiple PK sampling or other procedures to be performed in very tight timelines. Some protocols require an overnight stay and long-term stay, so, night/weekend shifts of doctors and nurses should be planned.
Specific requirements for Principal Investigator/Sub Investigator’s qualification in early phase trials should be followed per local legislation: e. g., medical license within the therapeutic area of the trial, previous experience in Phase I/Bioequivalence studies, or license of Clinical Pharmacologist.
Facilities and equipment
As facilities and equipment affect the progress of clinical trials, the Phase I unit should be located within a hospital environment with the critical care facilities available. For standalone units, 24/7 access to the hospital emergency response team should be available within minutes.
Appropriate emergency equipment should be available and procedures for handling medical emergencies should be in place. The staff should be trained in life support and how to use emergency equipment. Early phase units should be equipped with alarms in any areas where subjects have access (wards, showers, bathrooms, canteen, and recreational areas). Alarms should be regularly tested and the evaluation should be documented. One more important point to consider for early phase trials: synchronized clocks should be placed in procedure areas for accurate PK sampling, dosing, vital signs measurement.
Administration of healthy volunteers
Healthy volunteer identification and taking care of them is a third key factor in early phase units. To enroll the required number of subjects is a challenge in every clinical trial. The early phase units usually have their database of healthy volunteers. Three points to consider on the proper administration of healthy volunteers:
- A recruitment strategy should be developed for every trial separately with the prevention of “over-volunteering” considered.
- All advertisements for recruitment should be approved by the Ethics Committee before using them.
- The consenting of healthy volunteers for personal data processing should be properly documented to meet data protection requirements.
When a potential subject comes to the site, he/she should provide his/her passport or ID card. The site should have a procedure to check the subject’s identity for every visit to ensure the same subject has consented and the same subject is being dosed. Bracelets with subject ID number should be used during study visits, and the staff should check the subject’s ID before conducting any procedure.
Consenting procedure for healthy volunteers or patients in early phase trials is conducted per GCP guidelines and local regulations. Special attention should be paid to potential trial risks (especially in first-in-human trials), protocol compliance: e.g., long-term stays in the unit, restrictions for diet, smoking, alcohol consumption.
Compensation for participation in the trial is an additional topic during the informed consent procedure. The compensation scheme per local regulations should be approved by the Ethics Committee and documented in the Informed Consent Form.
A few more important things to consider for early phase units to ensure a safe and friendly environment: controlled access to the unit, locked windows, fire security system, and emergency exits, sufficient space for leisure, Wi-Fi, TV, books, magazines, board games, etc. And, of course, compliance with the protocol procedures, proper medical care, and protection of the subject’s rights is a benchmark for every clinical trial.
Here are outlined only a few points that should be considered for early phase units. Please refer to the international guidelines and local legislation for further guidance for these types of units.
Saulenė Kudarauskaitė, MD
Quality Assurance Manager
BIO1 Early Phase Unit
References:
- Annex V to Guidance for the conduct of Good Clinical Practice inspections. Phase I Units.
- MHRA Phase I Accreditation Scheme, Version 3.
"
["post_title"]=>
string(58) "
Quality Assurance in Early Phase Clinical Trials "
["post_excerpt"]=>
string(231) "Quality assurance in early phase clinical trials plays an important role in minimizing potential risk to volunteers or patients who take part in these types of studies and may help to eliminate potential problems before they occur."
["post_status"]=>
string(7) "publish"
["comment_status"]=>
string(4) "open"
["ping_status"]=>
string(4) "open"
["post_password"]=>
string(0) ""
["post_name"]=>
string(50) "quality-assurance-in-early-phase-clinical-trials-2"
["to_ping"]=>
string(0) ""
["pinged"]=>
string(0) ""
["post_modified"]=>
string(19) "2020-11-04 09:53:20"
["post_modified_gmt"]=>
string(19) "2020-11-04 09:53:20"
["post_content_filtered"]=>
string(0) ""
["post_parent"]=>
int(0)
["guid"]=>
string(29) "https://bio1trials.com/?p=549"
["menu_order"]=>
int(0)
["post_type"]=>
string(4) "post"
["post_mime_type"]=>
string(0) ""
["comment_count"]=>
string(1) "0"
["filter"]=>
string(3) "raw"
}
Quality Assurance in Early Phase Clinical Trials
Quality assurance in early phase clinical trials plays an important role in minimizing potential risk to volunteers or patients who take part in these types of studies and may help to eliminate potential problems before they occur.
Read more >
object(WP_Post)#1484 (24) {
["ID"]=>
int(516)
["post_author"]=>
string(1) "1"
["post_date"]=>
string(19) "2020-06-04 13:12:44"
["post_date_gmt"]=>
string(19) "2020-06-04 13:12:44"
["post_content"]=>
string(6754) "
Reimbursement for clinical trial participants is always a hot topic. Especially for healthy volunteers in early phase trials who do not get any additional benefits for participating in clinical trials and accept potential study risks. Monetary reimbursement is a strong motivator deciding whether to participate in a clinical trial or not. Evidence suggests that although there are often multiple motivations, healthy volunteers are primarily motivated by financial reward. Thus, it is not surprising that more money might influence their decisions.
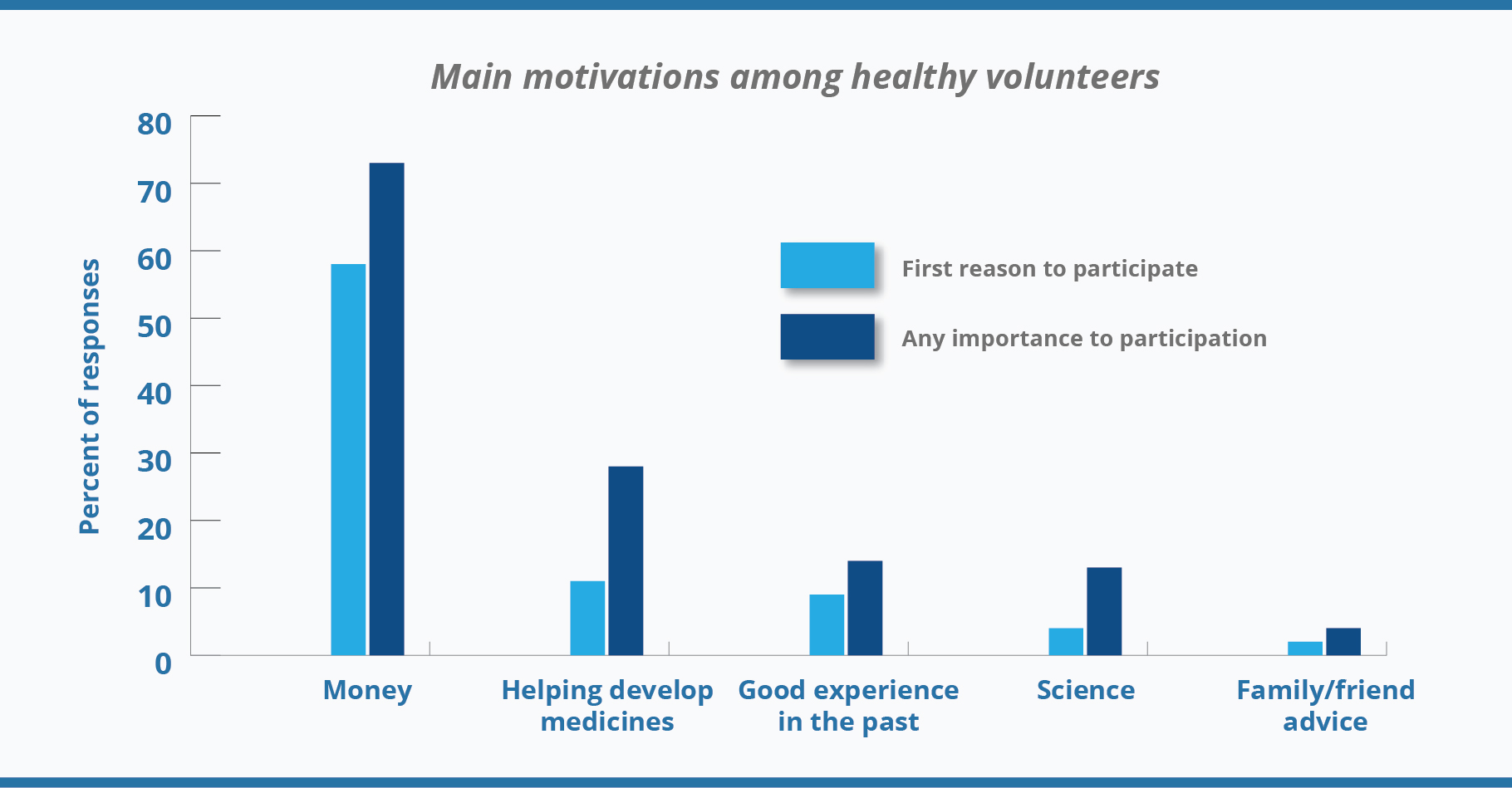
Although a financial reward is a primary motivation to participate in clinical trials for healthy volunteers, financial motivations are not the only ones. Contribution to science or the health of others, accessing ancillary healthcare benefits, scientific interest, or interest in the goals of the study, as well as meeting people and curiosity motivates the healthy volunteers too (
2). The diagram below demonstrates that not always money matter, some percentage of responders’ care about progress in medicine as well.
[caption id="attachment_517" align="aligncenter" width="1350"]
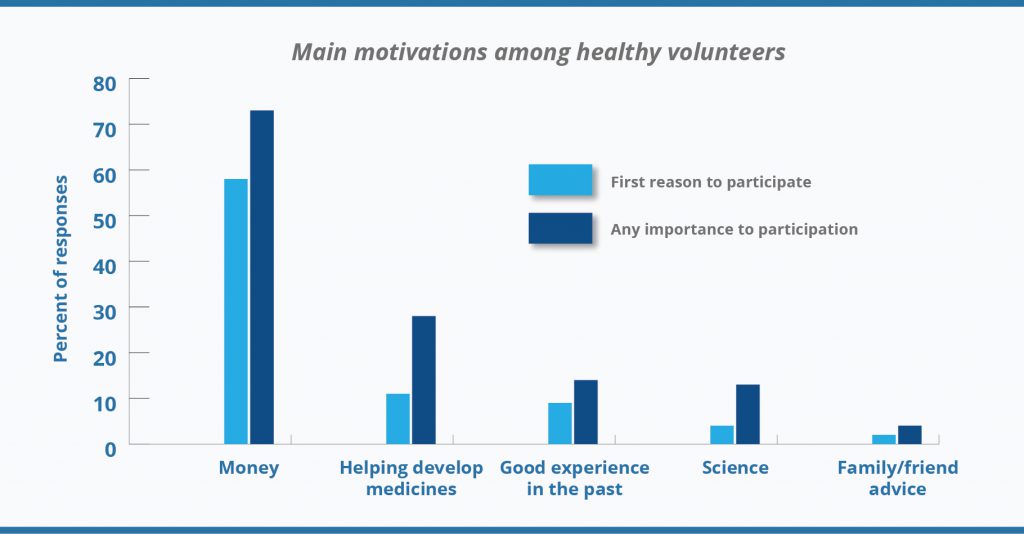
Source: Grady, C., Bedarida, G., Sinaii, N., Gregorio, M. A., & Emanuel, E. J. (2017). Motivations, enrollment decisions, and socio-demographic characteristics of healthy volunteers in phase 1 research. Clinical Trials, 14(5), 526–536.[/caption]
Legislation Changes in Lithuania
Let’s take a look at how legislation on reimbursement for clinical trial participants changed in Lithuania during the last five years.
The biggest changes in clinical trial reimbursement rules in Lithuania took place when a new revision of Law on Ethics of Biomedical Research entered into force on
01 January 2016 (
3). Please, see below the changes at a glance:

The amount of reimbursement is defined in Order No. V-15 (
08 January 2016) of the Minister of Health of the Republic of Lithuania (
4). According to this Order, the terms and conditions of reimbursement should be clearly outlined in a written agreement, i.e., in the approved Informed Consent Form. For the period between
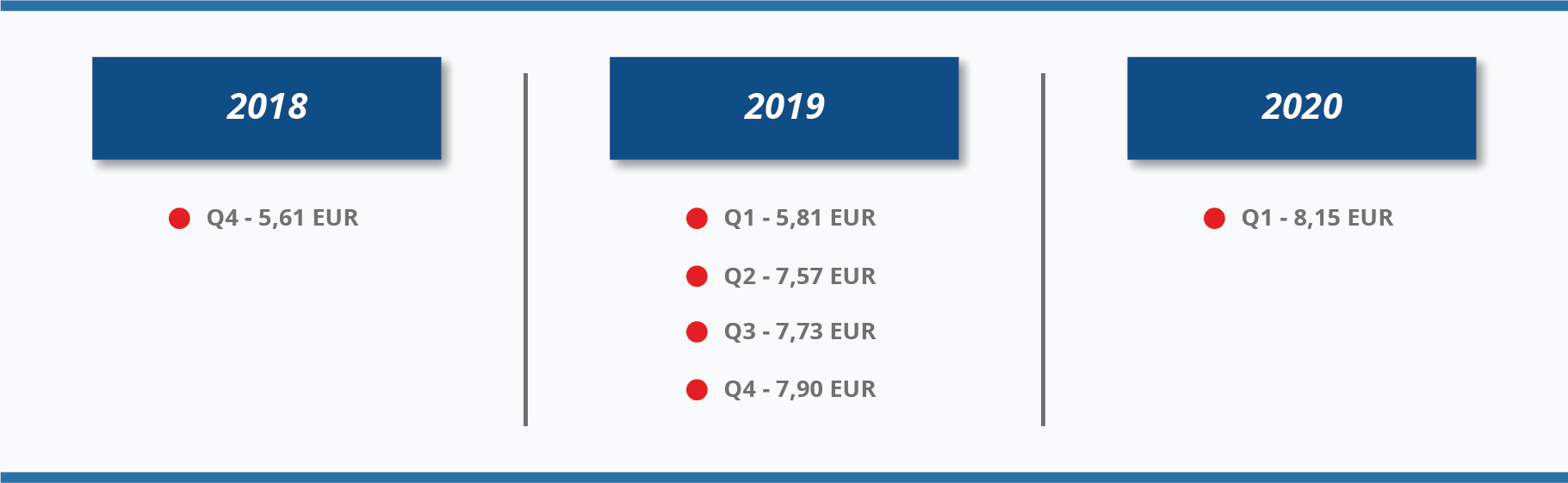
08 January 2016 and
11 September 2018 the amount of reimbursement for the time spent was calculated according to the minimum hourly earnings approved by the Government of the Republic of Lithuania, taking into account the time actually spent on biomedical research (time spent at the biomedical research centre and time for travelling), but not more than 8 hours a day. Since
11 September 2018, the reimbursement amount was increased up to the amount of average hourly gross earnings of the Republic of Lithuania (
5). Also, the cap of 8 hours per day was removed. You can see how the average maximum hourly reimbursement amount increased in 2018-2020 due to gross average monthly earnings growth:
 The amendment of Law on Ethics of Biomedical Research provided a better reimbursement scheme for trial participants since 01 January 2016. This is a fair and transparent way to reimburse clinical trial participants in Lithuania, especially, to healthy volunteers or subjects in early phase trials where some visits last up to two days or even longer. Thus, the changes in subject reimbursement scheme gave better chances to Lithuanian trial sites to be competitive in early phase trials and achieve recruitment targets.
The amendment of Law on Ethics of Biomedical Research provided a better reimbursement scheme for trial participants since 01 January 2016. This is a fair and transparent way to reimburse clinical trial participants in Lithuania, especially, to healthy volunteers or subjects in early phase trials where some visits last up to two days or even longer. Thus, the changes in subject reimbursement scheme gave better chances to Lithuanian trial sites to be competitive in early phase trials and achieve recruitment targets.
Saulenė Kudarauskaitė, MD
Quality Assurance Manager
BIO1 Early Phase Unit
References:
- Phase 1 healthy volunteer willingness to participate and enrollment preferences.
- More than the money: A review of the literature examining healthy volunteer motivations.
- Republic of Lithuania Law Amending Law No VIII-1679 on Ethics of Biomedical Research.
- Dėl Patirtų išlaidų ir sugaišto laiko dėl dalyvavimo biomedicininiame tyrime kompensacijos apskaičiavimo ir mokėjimo tvarkos aprašo patvirtinimo.
- Dėl Lietuvos Respublikos sveikatos apsaugos ministro 2016 m. sausio 8 d. įsakymo Nr. V-15 „Dėl Patirtų išlaidų ir sugaišto laiko dėl dalyvavimo biomedicininiame tyrime kompensacijos apskaičiavimo ir mokėjimo tvarkos aprašo patvirtinimo“ pakeitimo.
"
["post_title"]=>
string(74) "
Changes in Reimbursement of Clinical Trial Subjects in Lithuania "
["post_excerpt"]=>
string(230) "Reimbursement for clinical trial participants is always a hot topic. Especially for healthy volunteers in early phase trials who do not get any additional benefits participating in clinical trials and accept potential study risks."
["post_status"]=>
string(7) "publish"
["comment_status"]=>
string(4) "open"
["ping_status"]=>
string(4) "open"
["post_password"]=>
string(0) ""
["post_name"]=>
string(64) "changes-in-reimbursement-of-clinical-trial-subjects-in-lithuania"
["to_ping"]=>
string(0) ""
["pinged"]=>
string(0) ""
["post_modified"]=>
string(19) "2020-06-04 13:55:03"
["post_modified_gmt"]=>
string(19) "2020-06-04 13:55:03"
["post_content_filtered"]=>
string(0) ""
["post_parent"]=>
int(0)
["guid"]=>
string(29) "https://bio1trials.com/?p=516"
["menu_order"]=>
int(0)
["post_type"]=>
string(4) "post"
["post_mime_type"]=>
string(0) ""
["comment_count"]=>
string(1) "0"
["filter"]=>
string(3) "raw"
}
Changes in Reimbursement of Clinical Trial Subjects in Lithuania
Reimbursement for clinical trial participants is always a hot topic. Especially for healthy volunteers in early phase trials who do not get any additional benefits participating in clinical trials and accept potential study risks.
Read more >
object(WP_Post)#1483 (24) {
["ID"]=>
int(495)
["post_author"]=>
string(1) "1"
["post_date"]=>
string(19) "2020-04-23 08:30:57"
["post_date_gmt"]=>
string(19) "2020-04-23 08:30:57"
["post_content"]=>
string(7911) "
Highly-variable drugs (HVD) are the drugs whose within-subject variance is larger than 30% (1). In other words, most of the drugs have a predictable pharmacokinetic profile within a subject, but there are drugs whose pharmacokinetic profile may differ from one use to another. These drugs, with high within-subject variability, are believed to have a wide therapeutic window, and despite high variability, these products can be both safe and effective. However, with higher uncertainty around their pharmacokinetics and their efficacy, the chance of a spurious result increases. Therefore, it's important to take additional measures to account for the higher variance.
Under standard regulatory conditions for bioequivalence, HVD testing faces many challenges. In bioequivalence studies, where the 90% confidence interval for the ratio of the geometric means of the two drug products should be between 0.80 and 1.25
(2), the clinical trial would have to enrol an uncomfortably large number of subjects. This increases the costs as well as the chance of safety issues. For example, to demonstrate BE with 90% power, it was estimated that 136 subjects would be required for a drug with 60% within-subject coefficient of variation even if the test and reference products were identical
(3). To combat these issues, regulatory authorities introduced new procedures in the last decade.
The solution to higher within-subject variation is to scale the average bioequivalence with an intrasubject variation. Usually, this is done in two-period crossover studies from the residual variation. In investigations with three or more periods, where at least one formulation is administered twice, the within-subject variation of one or both formulations can be directly estimated
(2), The interpretation is quite similar to the model for average bioequivalence - the scaled confidence interval of average bioequivalence has to meet the limits preset by the regulatory bodies.
The limits differ between the FDA and EMA. The FDA suggests that the reference product should be measured at least twice in each subject. That means either a 3 or 4-period study is needed. The bioequivalence limit is scaled as well by a regulatory constant. FDA recommends 0.25 as the constant based on a 25% coefficient of variation. The scaling factor is used when the variance of the area under the curve contrasting plasma concentration with time (AUC) or the maximum concentration (Cmax) exceeds 30%. The confidence interval for BE acceptance scale accordingly (for mathematical details, read the computational procedure described by the FDA
(4)).
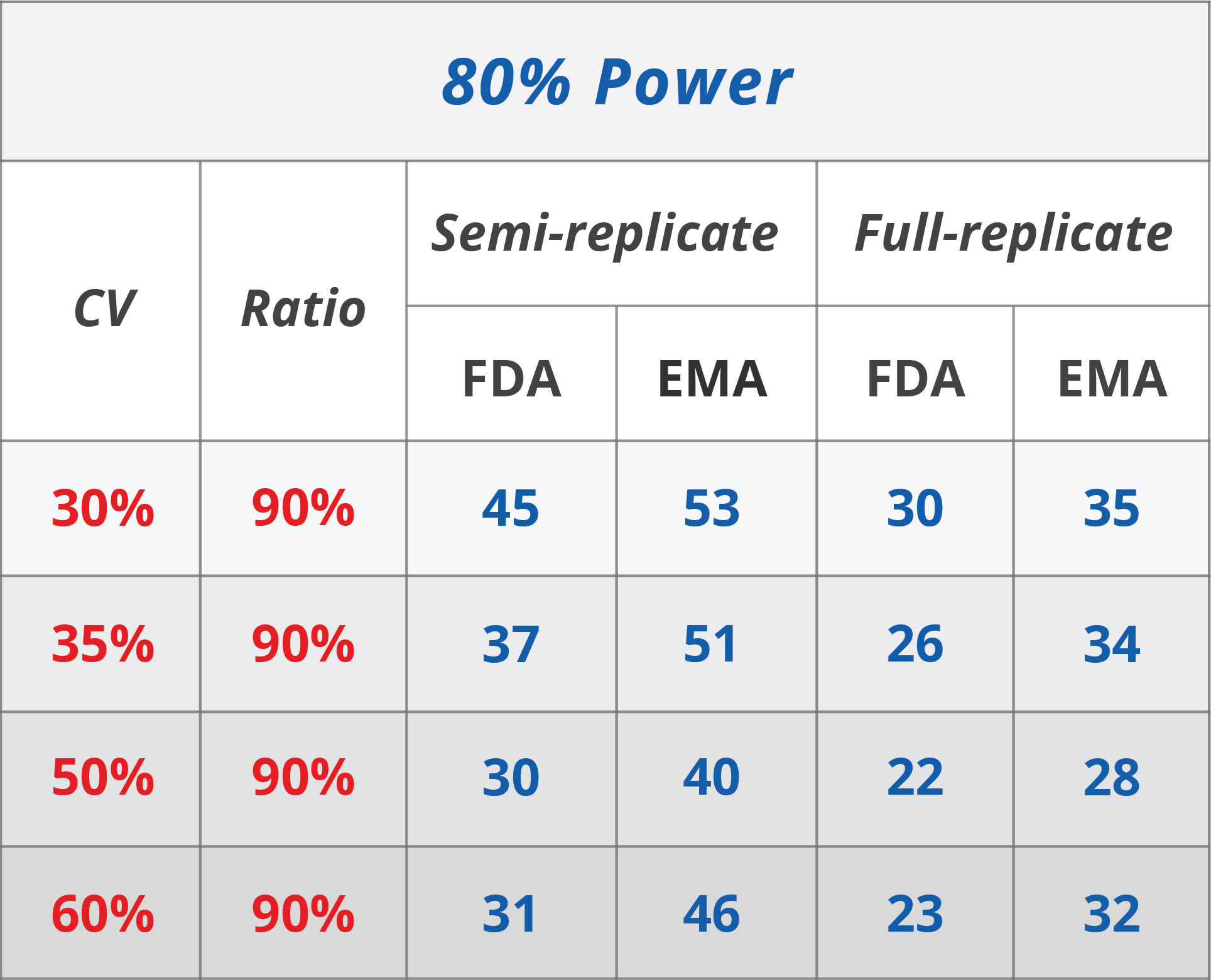
EMA also recommends the application of a mixed strategy, i.e. unscaled average bioequivalence should be applied when the intraindividual variation does not exceed 30%, but average bioequivalence with expanding limits (ABEL) should be used otherwise. However, EMA suggests an upper limit of 50% beyond which the bioequivalence limits remain constant and average bioequivalence is used without scaling again (See Table 1). EMA, notably, suggests the procedure be applied only to Cmax but not to AUC. EMA recommends a regulatory constant of 0.294 (corresponding to CV = 30%) and, therefore, 0.760 as the BE limit in the logarithmic scale.
[caption id="attachment_492" align="alignright" width="480"]
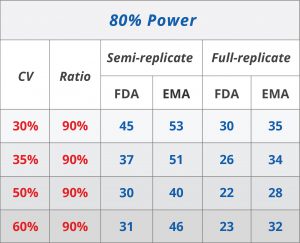
Table 1. Reference-scaled BE approach: Comparison of FDA and EMA (6).[/caption]
These differences do have important implications. Generally, FDA guidelines are more permissive and demonstrating BE is more likely under FDA guidelines rather than EMA requirements. Coming back to the regulatory constants, EMA uses 0.294 when the intrasubject CV is larger than 30%, though, FDA uses 0.25 at the same threshold. Whereas the bioequivalence limits under the EMA framework scale gradually, a discontinuity is introduced using the FDA’s regulatory coefficient.
This has important implications for the consumer as well. Type I error, the probability of rejecting a null hypothesis when it is, in fact, correct, in this case, means that two products are accepted as bioequivalent when they are, in fact, not bioequivalent. This type of error is also known as consumer risk. The error is kept small, and sample sizes are used to keep it at 5%. However, under the mixed strategy framework, the observed variance of a drug may differ from the true variance. As an example, if the observed variance is over 30%, but the true variance is 29%, the drug would be considered highly variable, and wider acceptance limits would be applied. Because of this, there is a higher risk of false acceptance. The type I error estimates vary, but the EMA regulatory constant is more favourable and reduces the risk that a non-bioequivalent drug will reach the consumer market.
The approach of scaled bioequivalence is enormously advantageous: it decreased the number of required subjects and allowed testing highly variable drugs. In principle, both the FDA and EMA have similar approaches to scaling bioequivalence, but the differences favour the approach suggested by EMA. There is still much work to be done to account for higher than nominal Type I errors as well as revisiting the optimal study design under the FDA guidelines.
Simonita Maciulskyte, MD
Clinical Research Physician
BIO1 Early Phase Unit
References:
- Guideline on The Investigation of Bioequivalence.
- Bioequivalence for highly variable drugs: regulatory agreements, disagreements, and harmonization.
- Bioequivalence Approaches for Highly Variable Drugs and Drug Products.
- Draft Guidance on Progesterone.
- Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs - General Considerations.
- Tothfalusi, J. Pharm.Pharmaceutic Sci. 15 (1) 73-84, 2012.
"
["post_title"]=>
string(78) "
Bioequivalence Studies and Highly Variable Drugs: Things to Consider "
["post_excerpt"]=>
string(248) "Highly-variable drugs (HVD) are the drugs whose within-subject variance is larger than 30%. In other words, most of the drugs have a predictable pharmacokinetic profile within a subject, but there are drugs whose pharmacokinetic profile may differ."
["post_status"]=>
string(7) "publish"
["comment_status"]=>
string(4) "open"
["ping_status"]=>
string(4) "open"
["post_password"]=>
string(0) ""
["post_name"]=>
string(67) "bioequivalence-studies-and-highly-variable-drugs-things-to-consider"
["to_ping"]=>
string(0) ""
["pinged"]=>
string(0) ""
["post_modified"]=>
string(19) "2020-04-24 11:41:38"
["post_modified_gmt"]=>
string(19) "2020-04-24 11:41:38"
["post_content_filtered"]=>
string(0) ""
["post_parent"]=>
int(0)
["guid"]=>
string(29) "https://bio1trials.com/?p=495"
["menu_order"]=>
int(0)
["post_type"]=>
string(4) "post"
["post_mime_type"]=>
string(0) ""
["comment_count"]=>
string(1) "0"
["filter"]=>
string(3) "raw"
}
Bioequivalence Studies and Highly Variable Drugs: Things to Consider
Highly-variable drugs (HVD) are the drugs whose within-subject variance is larger than 30%. In other words, most of the drugs have a predictable pharmacokinetic profile within a subject, but there are drugs whose pharmacokinetic profile may differ.
Read more >
object(WP_Post)#1452 (24) {
["ID"]=>
int(379)
["post_author"]=>
string(1) "1"
["post_date"]=>
string(19) "2019-09-05 07:21:49"
["post_date_gmt"]=>
string(19) "2019-09-05 07:21:49"
["post_content"]=>
string(3141) "[caption id="attachment_380" align="alignleft" width="498"]
 Justinas Ivaška, General Manager of BIO1
Justinas Ivaška, General Manager of BIO1[/caption]
September 2019 – Pioneering Phase I unit in the Baltic States, BIO1, officially announced the appointment of Justinas Ivaška to the role of General Manager.
Located in the leading university hospital in Lithuania from primary care to tertiary level - Vilnius University Hospital,
Santaros Klinikos, BIO1 creates an outstanding space to reach added value in clinical trials. As the first early phase unit in the region, BIO1 is dedicated to ensuring the highest standards when working with healthy volunteers and patients.
Justinas Ivaška will play an important role in the provision of early phase unit services, ensuring highest quality standards and maximizing the efficiency of clinical conduct. Justinas will be responsible for daily operations and will work on the implementation of the overall company business development strategy.
„Justinas combines the in-depth medicinal knowledge with experience of business development and management. His professional background reveals that Justinas is a decisive strategic and analytical leader, what’s make him exceptionally well suited to lead BIO1 improvement. Justinas undoubtedly will be a highly valued addition to BIO1 team.“ said
Eglė Pavydė, BIO1 Director Business Development.
Justinas Ivaška graduated from Vilnius University, Faculty of Medicine, qualified as Medical Doctor. As well, Justinas holds a Bachelor degree in Business administration and Management and a PhD degree in Biomedical Sciences. Since 2017, Justinas is an Associate professor of the Faculty of Medicine at Vilnius University. Justinas Ivaška has more than 21 years of experience working as a medical doctor, specializing in otorhinolaryngology. Before joining BIO1, Justinas was working as Director of Business Development at Vilnius University Hospital, Santaros Klinikos. He was responsible for project management, the establishment of new health care departments, medical systems and implementation of therapeutic technologies.
About BIO1 Clinical Trial Unit
BIO1 provides facilities and expertise to investigate novel medicines and medical devices. We occupy a large clinical space of 500 m2 plus an administrative area of 120 m2 – all of which is modern, immaculate and has the very latest equipment. Combining the knowledge with the best doctors in the region, we ensure the highest standards in early phase clinical trials."
["post_title"]=>
string(39) "
BIO1 appoints General Manager "
["post_excerpt"]=>
string(159) "September 2019 – Pioneering Phase I unit in the Baltic States, BIO1, officially announced the appointment of Justinas Ivaška to the role of General Manager."
["post_status"]=>
string(7) "publish"
["comment_status"]=>
string(4) "open"
["ping_status"]=>
string(4) "open"
["post_password"]=>
string(0) ""
["post_name"]=>
string(29) "bio1-appoints-general-manager"
["to_ping"]=>
string(0) ""
["pinged"]=>
string(0) ""
["post_modified"]=>
string(19) "2019-09-09 18:53:24"
["post_modified_gmt"]=>
string(19) "2019-09-09 18:53:24"
["post_content_filtered"]=>
string(0) ""
["post_parent"]=>
int(0)
["guid"]=>
string(28) "https://bio1trials.com/?p=379"
["menu_order"]=>
int(0)
["post_type"]=>
string(4) "post"
["post_mime_type"]=>
string(0) ""
["comment_count"]=>
string(1) "0"
["filter"]=>
string(3) "raw"
}
BIO1 appoints General Manager
September 2019 – Pioneering Phase I unit in the Baltic States, BIO1, officially announced the appointment of Justinas Ivaška to the role of General Manager.
Read more >